Mount Sinai scientists, in collaboration with researchers from the Carlos III Health Institute (ISCIII) in Madrid, Spain, have located and identified alterations in the monkeypox virus genome that potentially correlate with changes in the virus’s transmissibility observed in the 2022 outbreak. The findings were published April 18 in Nature Communications.
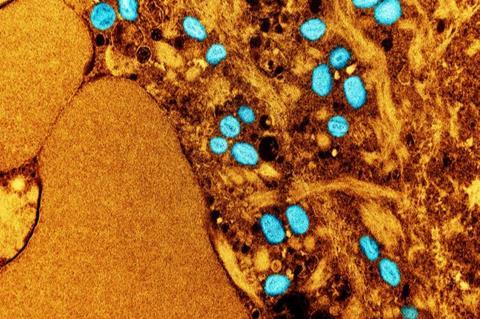
Monkeypox virus (MPXV) is a double-stranded DNA virus that can infect animals and humans. MPXV causes a disease known as mpox, with symptoms that include fever, swollen lymph nodes, and a rash. Most cases are mild and tend to get better on their own; however, mpox can be very painful and may lead to permanent scarring.
First encountered in 1958 in crab-eating macaque monkeys imported to Belgium, MPXV has caused sporadic human disease outbreaks since the 1970s in Central and Western Africa. In May 2022, multiple countries, including the United States, reported an increasing number of MPXV infections and associated disease, including clusters in cases potentially linked to super-spreading events in Belgium, Spain, and the United Kingdom.
While the number of new cases associated with the 2022 spillover has decreased over time, cases of the disease are still occurring among unvaccinated individuals, including a current increase in Central Africa due to a new spillover. As the virus’s circulation in humans increases, the risk of emergence of a more transmissible variant capable of becoming endemic in the human population increases.
Clear alert signals
“Biopreparedness and virological surveillance involves studying the causes that favor zoonotic spillover and facilitates human-to-human transmission. When we observe significant changes in basic epidemiological features of a viral agent like monkeypox, it should reinvigorate our interest in understanding those transmission conditions. The increasing number of cases currently happening in Africa, and the 2022 epidemic, should be clear alert signals,” says Gustavo Palacios, PhD, Professor of Microbiology at the Icahn School of Medicine at Mount Sinai and a senior author of the study.
To carry out the study, researchers analyzed samples from 46 patients infected with MPXV whose diagnosis and sequencing were carried out at the ISCIII at the beginning of the 2022 mpox outbreak. The team performed high-quality sequencing of each study participant’s complete monkeypox virus genome to determine possible correlations between genomic variations in the different groups of sequences and epidemiological links associated with the virus’s ability to evolve, transmit, and infect.
Genomic changes
According to the research team, recurrent observed genomic changes were located in areas of the genome that could be related to viral adaptation. Those specific locations would contribute to modulating the viral replication cycle, adaptability, and path of entry and egress. These alterations appear in areas known as low complexity genomic regions, which are particularly difficult to sequence and analyze, explaining why they were overlooked before. This highly sophisticated complete genome sequencing was made possible through the use of two advanced sequencing technologies: single-molecule long-read sequencing (to cover highly repetitive regions) and deep short sequencing reads (to provide accuracy and depth).
By detailing the genomic alterations within these repetitive genomic sequences and linking them to critical viral functions, researchers provide a plausible explanation for the heightened transmissibility observed during the 2022 mpox outbreak.
“These findings might be offering the first hints to help us understand the unique features of the strains associated with sustained human-to-human transmission, which has not ever been observed in these agents,” says Dr. Palacios. “Better understanding of the doors that facilitate transmission of viral agents and impact their clinical presentations will enable us to develop more effective prevention and treatment strategies.”


No comments yet