A team led by CUNY Graduate Center biologists has produced a genetic analysis of Lyme disease bacteria that may pave the way for improved diagnosis, treatment, and prevention of the tick-borne ailment.
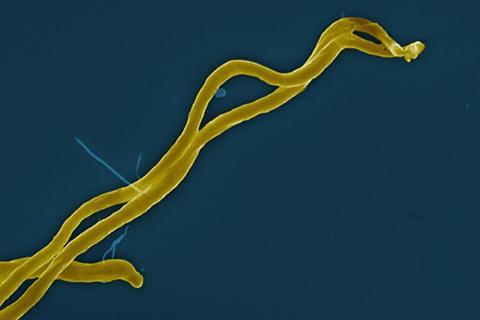
The international team mapped the complete genetic makeup of 47 strains of Lyme disease-related bacteria from around the world, creating a powerful tool for identifying the bacterial strains that infect patients. Researchers said this could enable more accurate diagnostic tests and treatments tailored to the bacteria causing each patient’s illness.
READ MORE: ‘Subway map’ of Lyme disease pathways IDs potential treatment targets
READ MORE: The climate crisis and the spread of vector-borne disease
“By understanding how these bacteria evolve and exchange genetic material, we’re better equipped to monitor their spread and respond to their ability to cause disease in humans,” said Weigang Qiu, a professor of Biology at the CUNY Graduate Center and Hunter College, and the corresponding author of the study.
The study was published in mBio journal.
Path to vaccinations
Researchers said the genetic information uncovered in the study may help scientists develop more effective vaccines against Lyme disease.
Lyme disease is the most common tick-borne illness in North America and Europe, affecting hundreds of thousands of people a year. The disease arises from bacteria belonging to the Borrelia burgdorferi sensu lato group, which infect people through the bite of infected ticks. Symptoms can include fever, headache, fatigue, and a characteristic skin rash. If left untreated, the infection can spread to joints, the heart, and the nervous system, causing more severe complications.
Case numbers are increasing steadily, with 476,000 new cases each year in the United States, and may grow faster with climate change, the authors of the study said.
Complete genomes
The research team, led by scientists from the CUNY Graduate Center and Hunter College, Rutgers, Stony Brook, and more than a dozen other research institutions, sequenced the complete genomes of Lyme disease bacteria representing all 23 known species in the group. Most hadn’t been sequenced before the effort. The National Institutes of Health-funded project included many bacteria strains most associated with human infections and species not known to cause disease in humans.
By comparing these genomes, the researchers reconstructed the evolutionary history of Lyme disease bacteria, tracing the origins back millions of years. They discovered the bacteria likely originated before the breakup of the ancient supercontinent Pangea, explaining the current worldwide distribution.
The study also disclosed how these bacteria exchange genetic material in and between species. This process, known as recombination, allows the bacteria to rapidly evolve and adapt to new environments. The researchers identified specific hot spots in the bacterial genomes where this genetic exchange occurs most frequently, often involving genes that help the bacteria interact with their tick vectors and animal hosts.
Software tools
To facilitate ongoing research, the team has developed web-based software tools (BorreliaBase.org) that allow scientists to compare Borrelia genomes and identify determinants of human pathogenicity.
Looking ahead, the researchers said they plan to expand their analysis to include more strains of Lyme disease bacteria, especially from understudied regions. They also aim to investigate the functions of genes unique to disease-causing strains, which could uncover new targets for therapeutic interventions. As Lyme disease expands its geographic range because of climate change, the research provides valuable tools and insights for combating this rising public health threat.
The study is supported by grants from NIH and an award from the Steven and Alexandra Cohen Foundation.





No comments yet