Following on from the recent official launch of BGI’s new CycloneSEQ™ is the first independently reviewed benchmarking data. The CycloneSEQ™ platform delivers long-reads using novel nanopore technology.
A new study tests the performance of a new platform in sequencing diverse microbial genomes and presents both the raw and processed data to allow others to scrutinize and verify the work. To provide further transparency, the peer-reviews, scripts and protocols of this benchmarking process are also included, allowing others to directly assess this new technology.
READ MORE: Healthy gut bacteria that feed on sugar analyzed for the first time
By directly reading DNA molecules without fragmentation, the analysis demonstrates that CycloneSEQ delivers long-read data with impressive length and accuracy, unlocking gaps that short-read technologies alone cannot bridge. This work has been published in the open-science journal GigaByte.
Filling the gaps in millions of bacterial genomes
While long established short-read sequencing technology remains cost-effective and accurate, struggles to assemble complete, circular bacterial genomes remain—leaving critical gaps in our understanding of microbial functions. The researchers from BGI Research have combined CycloneSEQ long-reads with DNBSEQ™ short-reads, achieving closed, circular, high-accuracy genomes for common gut bacteria.
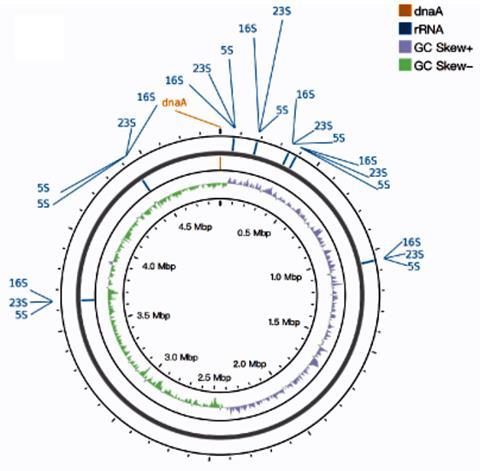
As a standard validation approach, the researchers sequenced the bacterial reference strain Akkermansia muciniphila ATCC BAA-835, yielding 12.07 Gbp of long-reads with an average length of 11.6 kbp. The hybrid assembly method outperformed both short- and long-read-only approaches, successfully assembling a complete genome of ATCC BAA-835 with a mismatch rate lower than 0.0001%.
The researchers subsequently sequenced 10 common strains isolated from the human gut and successfully closed all 10 bacterial genomes, as well as tiny phage/plasmid circles typically missed by short reads alone. Using long-read-only assemblies, they managed to put together complete, circularized genomes for 8 out of 10 strains, whereas using the previous generation short-read-only assembly failed to produce a single complete genome.
Repetitive regions
The researchers also revealed that the issue was not the inability of short-read sequencing to detect relevant regions but rather the inherent limitations of short-read assembly, typically caused by the presence of repetitive regions and high GC content in these genomes.
The technology also was tested using complex microbial communities. In a 21-strain synthetic gut community (18 bacteria, 2 fungi, 1 archaeon strains), hybrid assembly using CycloneSEQ + DNBSEQ outperformed some other single-method approaches, yielding 5 complete metagenome-assembled genomes (MAGs)—which was not achieved by short- or long-read assemblies alone.
CycloneSEQ’s long-reads currently deliver sufficient length and quality for circular genome assembly, but the using short-reads remains essential for polishing accuracy. The researchers future work is aimed assessing and using non-synthetic samples, fine-tuning the balance between short-read and long-read data to provide even faster, higher-quality assemblies.





No comments yet